代谢重编程是肿瘤的主要特征之一。肿瘤细胞通过改变自身的代谢途径或方式来满足其快速生长所需要的能量和代谢前体物质。金沙js4399首页江鹏课题组近期的研究发现:尿素循环、多胺合成和天冬酰胺代谢的异常改变可帮助肿瘤细胞,特别是p53缺失或突变的肿瘤细胞的增殖或存活(Li et al, Nature, 2019;Deng et al, Nature Communication, 2020)。相较于野生型的细胞,p53缺失或突变的肿瘤细胞是否对某种代谢途径产生特异的依赖,一直是领域内非常引人关注的重要问题之一。2013年来自英国Karen Vousden教授课题组的一项重要研究表明,serine缺乏会对p53缺失的肿瘤细胞造成致命性的伤害,而野生型细胞可通过代谢改变和细胞周期调控而存活下来(Maddocks et al., Nature, 2013)。
2021年7月10日,金沙js4399首页江鹏课题组在《美国科学院院报》(PNAS)发表了题为“p53缺陷通过诱导MTHFD2转录上调促进细胞增殖和抑制DNA损伤”(p53 deficiency induces MTHFD2 transcription to promote cell proliferation and restrain DNA damage)的研究论文。本项研究揭示了p53缺失或突变肿瘤细胞对一碳代谢酶MTHFD2的极端依赖性。p53的缺失或突变会直接导致MTHFD2的转录上调,并增高线粒体一碳代谢通路的活性。MTHFD2的高表达,一方面为p53缺失或突变的肿瘤细胞提供了合成嘌呤核苷酸所需的一碳单位,另一方面显著降低了细胞内活性氧的水平,增强了细胞的抗氧化能力。
此外,深入的机制研究发现,MTHFD2具有代谢非依赖的功能特性。MTHFD2在细胞核内可与PARP3结合,增强肿瘤细胞内非同源末端连接 (NHEJ)介导的DNA损伤修复能力,导致p53的缺失或突变肿瘤细胞对MTHFD2的抑制,以及对化疗药物Doxorubicin的敏感性大大提高。而在p53野生型肿瘤细胞中, 抑制MTHFD2会引起AICAR的累积,并激活了AMPK-p53-p21通路,从而维持基因组稳定性,最终帮助细胞存活下来(图1)。
因此,该项研究发现了MTHFD2高表达可能是p53缺失或突变肿瘤细胞的重要的代谢特征和关键的代谢脆弱点。相较于p53野生型肿瘤细胞,p53缺失或突变肿瘤细胞对于MTHFD2的抑制更加敏感,为临床治疗p53缺失或突变肿瘤提供了潜在的药物靶点。
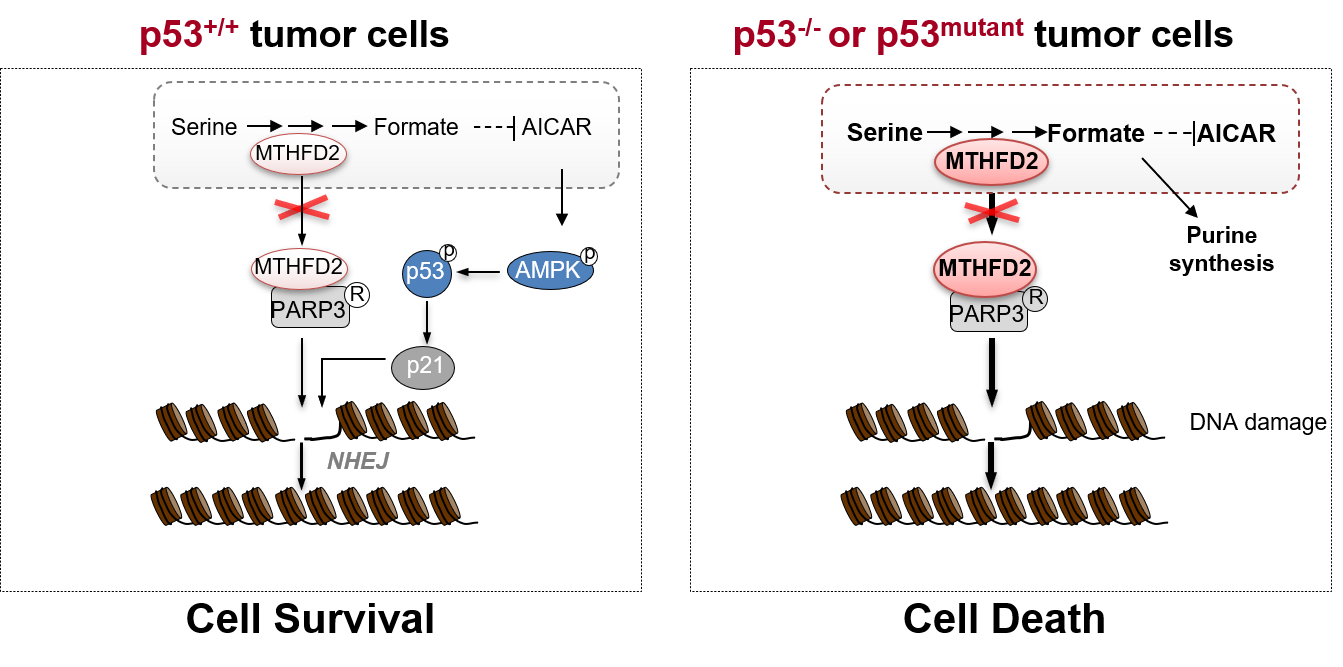
图1. p53缺失或突变的肿瘤细胞因MTHFD2的高表达而拥有异常增高的一碳代谢活性和DNA损伤修复能力,从而使其对MTHFD2产生极端的依赖。
金沙js4399首页生命学院江鹏实验室李根博士和吴珺博士为本文的共同第一作者。金沙js4399首页生命学院江鹏研究员为论文通讯作者。该项研究得到了清华-北大生命科学联合中心(CLS)和国家自然科学基金委经费的支持。
原文连接:https://www.pnas.org/content/118/28/e2019822118/tab-figures-data

